Arch Iran Med. 28(7):387-397.
doi: 10.34172/aim.34187
Original Article
Investigation of the Clinical and Genetic Spectrum of PMM2-CDG: Insights from a Family with a Novel Variant and Previous Studies
Parnian Alagha Conceptualization, Data curation, Formal analysis, Project administration, Software, Writing – original draft, Writing – review & editing, 1, # 
Tara Akhtarkhavari Data curation, Formal analysis, Project administration, Software, Writing – original draft, Writing – review & editing, 1, #
Ebrahim Shokouhian Investigation, Methodology, Visualization, 1
Fatemeh Ghodratpour Investigation, Methodology, Visualization, 1
Sanaz Arzhangi Formal analysis, Visualization, 1
Hossein Najmabadi Resources, Supervision, Validation, 1, *
Kimia Kahrizi Funding acquisition, Resources, Supervision, Validation, 1, *
Author information:
1Genetics Research Center, University of Social Welfare and Rehabilitation Sciences, Tehran, Iran
#Joined first authors.
Abstract
Background:
PMM2-CDG, also known as congenital disorder of glycosylation type 1a, is the most common N-linked glycosylation disorder, characterized by a wide range of neurological and multisystem manifestations. Understanding the genotype-phenotype correlations is essential for accurate diagnosis and patient management. This study aims to identify the genetic cause of PMM2-CDG in an Iranian family with multiple affected members, and to analyze the genetic and clinical spectrum of the disorder through a comprehensive literature review.
Methods:
Exome sequencing re-analysis was performed to detect disease-causing variants in three affected siblings. Additionally, a literature review was conducted, analyzing 91 previously reported cases of PMM2-CDG to determine the most prevalent variants and associated clinical features.
Results:
A novel splice site variant (c.640-9T>A) was identified alongside a previously reported missense mutation (c.647A>T; p.N216I) in the affected individuals. The literature review revealed that the most frequent PMM2 variants were p.R141H (28.8%), p.V231M (12.8%), p.N216I (6.4%), and p.V129M (5.8%), with 77.6% of mutations occurring in exons 5 and 8. The most common clinical findings included developmental delay, ocular abnormalities (hypertelorism, strabismus), muscular system defects (hypotonia, muscle weakness), neurological symptoms (abnormal MRI findings), cardiovascular involvement (pericarditis, pericardial effusion), and clotting disorders.
Conclusion:
We expect that our detailed clinical study will improve the genotype-phenotype interpretation of causal PMM2-CDG variants and the analysis of next-generation sequencing data, leading to clarification of the cause of complicated cases of rare diseases.
Keywords: Congenital disorder of glycosylation type 1A, Genotype-phenotype correlation, Novel variant, PMM2 gene, Novel variant
Copyright and License Information
© 2025 The Author(s).
This is an open-access article distributed under the terms of the Creative Commons Attribution License (
https://creativecommons.org/licenses/by/4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Cite this article as: Alagha P, Akhtarkhavari T, Shokouhian E, Ghodratpour F, Arzhangi S, Najmabadi H, et al. Investigation of the clinical and genetic spectrum of PMM2-CDG: insights from a family with a novel variant and previous studies. Arch Iran Med. 2025;28(7):387-397. doi: 10.34172/aim.34187
Introduction
Congenital disorders of glycosylation (CDGs) constitute a heterogeneous group of inherited metabolic diseases characterized by defects in glycoprotein and glycolipid glycan synthesis and attachment. Over 160 CDG subtypes have been described,1,2 encompassing N-linked, O-linked, and hybrid N- and O-linked glycosylation, as well as lipid and glycosylphosphatidylinositol (GPI) anchor biosynthesis abnormalities.3 The first CDG was identified by Jaeken and colleagues in 1980, affecting approximately 1 in 20 000 individuals.3-5
Clinically, patients often present with a recognizable phenotype characterized by neurological and multisystem manifestations, which can complicate the diagnosis of CDG. The severity of PMM2-CDG varies widely, ranging from severe neonatal forms with a high mortality rate (approximately 20% within the first year of life) to milder presentations in adulthood.5-7 Neurological signs are the primary clinical feature of PMM2-CDG, affecting both the central and peripheral nervous systems. These neurological abnormalities may occur alone or alongside systemic abnormalities.2
During infancy, the affected individuals frequently present with neurological deficits, including cerebellar hypoplasia, hypotonia, ataxia, and hyporeflexia, as well as strabismus. Additionally, failure to thrive, hepatic problems, and developmental delay are commonly observed.3,8 Hypotonia, ataxia, retinitis pigmentosa, seizure, intellectual disability (IQ 40‒70), stroke-like episodes, speech and movement impairments, peripheral neuropathy, coagulopathy, and skeletal abnormalities are common features in the affected children.3 Retinitis pigmentosa, myopia, joint contractures, non-progressive cognitive dysfunction, and peripheral neuropathy are common clinical findings in adolescents with PMM2-CDG.9
PMM2-CDG results from mutations in the PMM2 gene, located on chromosome 16p13.2, which encodes a 246-amino acid protein. This gene is broadly expressed in both human and mouse tissues.10 As a member of the HAD-IIB phosphomutase subfamily within the larger HAD superfamily of hydrolases, PMM2 possesses a conserved alpha/beta core domain, a structural feature shared among homologs spanning bacteria, archaea, and eukaryotes.11 Structurally, PMMs are composed of a core domain (residues 1‒90 and 198‒262) that houses the active site with four conserved motifs, and a cap domain (residues 95‒194), which plays a role in enzyme function and stability.10,12,13
PMM2 encodes a homodimeric cytosolic isomerase that catalyzes the conversion of mannose-6-phosphate to mannose-1-phosphate in the cytosol, with glucose 1,6-bisphosphate serving as an activator.3,5,14 Mannose-1-phosphate is an essential precursor for synthesizing GDP-mannose and dolichol-phosphate-mannose, both of which serve as mannose donors in N-linked glycosylation pathways.5,15,16 N-linked glycosylation is an important post-translational modification that involves a variety of processes including protein folding, signaling, trafficking, protein stability, localization, cell adhesion, etc.17,18 In addition to its extensive role in cellular functions, the importance of PMM2 is highlighted by a study demonstrating that targeted disruption of the PMM2 gene in mice results in early embryonic lethality.10,19
Enzymes responsible for catalyzing N-glycosylation are ubiquitously expressed throughout both developing and adult nervous tissue.18 Studies have demonstrated the vital role of N-glycosylation in both neurodevelopmental processes and the functioning of the mature brain.20 N-glycosylation is essential for neuronal function, influencing various cell types including neurons, astrocytes, and microglia.20 Furthermore, fucosylated glycans, synthesized from GDP-mannose,21 play a crucial role in cognitive processes such as learning and memory.22 These findings align with the clinical observation that almost all patients with PMM2-CDG exhibit neurological symptoms.
Here, we describe an Iranian family with three individuals affected by the rare congenital disorder of glycosylation type 1a, who have a compound heterozygote variant in the PMM2 gene; it is the third family with PMM2-CDG reported from Iran with a new nucleotide substitution. Our results further underscore the importance of a thorough and systematic re-evaluation of phenotypic descriptions, alongside using an up-to-date and reviewed pipeline for reanalysis of WES data.
Materials and Methods
An Iranian family (from Babol city, northern Iran) was previously referred to the Genetics Research Center (GRC) of the University of Social Welfare and Rehabilitation Sciences (USWR) for genetic investigation of intellectual disability, but the pathogenic variant(s) were not identified in our previous NGS investigation of the family. We performed a re-analysis study on the family, clinical re-examinations were conducted for affected individuals by a specialist clinician, and the clinical records were reviewed. Written informed consent was obtained from the parents of the patients and normal siblings. The study was approved by the Ethics Committee of the University of Social Welfare and Rehabilitation Sciences, Tehran, Iran. Peripheral blood samples were collected and genomic DNA was extracted using the salting-out protocol.
Samples collected from the proband of the family underwent re-sequencing using the Agilent SureSelectXT Human All Exon V6 Kit (Agilent Technologies, Santa Clara, CA, USA), and sequencing was performed on Illumina NextSeq500 (Illumina, San Diego, CA, USA). Since the GATK platform was used in the previous analysis, in this re-analysis, in addition to aligning raw sequenced data against the human reference genome hg38/GRCh38, sorting, duplicate marking, base quality recalibration, and small variant SNV and indel calling were performed using the Illumina DRAGEN Bio-IT Platform V3. The generated VCF file was uploaded to Ilyome (https://www.ilyome.com) for re-annotation and re-analysis. Variant filtering on the Ilyome platform was conducted by considering their quality (depth greater than 3) and allele frequency (less than 1% in gnomAD genomes, gnomAD exomes, Genoks, 1000 genomes, and TOPMED bravo databases). In the next step of variant filtering, variants including stop gained, frameshift, stop or start lost, transcript amplification, in-frame insertion and deletion, missense, protein-altering, and splice region variants with good coverage would remain for analysis. Variants were prioritized based on variant impact, inheritance patterns, phenotype compatibility, allele count in population databases (allele count for homozygous and heterozygous was 0 and less than 10, respectively), and in-silico prediction scores. Additionally, the analysis of variants involved the utilization of various databases, including Online Mendelian Inheritance in Man (OMIM, https://www.omim.com), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar), Varsome (https://varsome.com), Franklin (https://franklin.genoox.com), and PubMed, as well as in-silico prediction tools such as SIFT, MutationTaster, REVEL, MetaRNN, CADD, dbscSNV, and SpliceAI (for splicing variants).
Furthermore, we conducted a comprehensive review of published studies reporting PMM2 variants and associated clinical data from 2017 to 2024 as this is a descriptive study aimed at investigating additional genotype-phenotype correlations. PubMed and Google Scholar were used as primary databases for this review. We excluded papers that reported solely clinical data, exclusively molecular data, or provided cohort-level data without individual patient clinical information.
Results
Family History and Clinical Presentation
The family had three affected siblings; two females (aged 61 and 46 years) and one male (aged 68 years), born to unrelated parents but originating from the same village. All presented with developmental delay, microcephaly, severe intellectual disability, strabismus, short stature (150 cm, 130 cm, and 145 cm, respectively), hypotonia, and spastic paraplegia. Hearing impairment and seizures were absent. The pedigree of the family is depicted in Figure 1A.
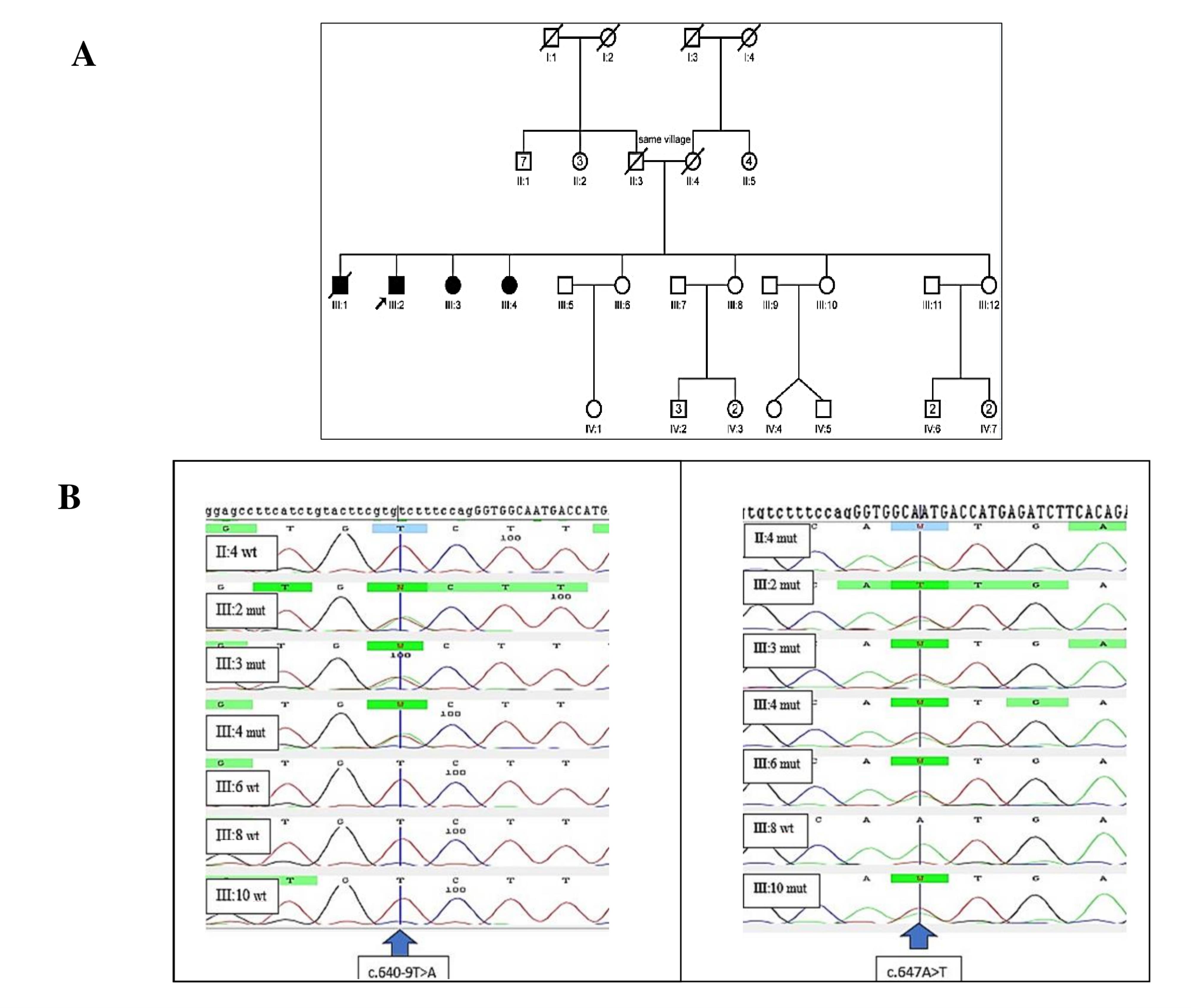
Figure 1.
(A) Pedigree of the family. (B) Sanger sequencing of the c.647A > T and c.640-9T > A variants in the family. Wild type (wt) and mutated sequences; Arrow shows the mutated position
.
(A) Pedigree of the family. (B) Sanger sequencing of the c.647A > T and c.640-9T > A variants in the family. Wild type (wt) and mutated sequences; Arrow shows the mutated position
Patients’ Genotypes and Variant Description
Our study revealed compound heterozygous variants in the three affected siblings, comprising a previously reported missense mutation c.647A > T (p.N216I) and a novel splice site variant (c.640-9T > A) of uncertain significance. The electropherogram of c.640-9T > A and c.647A > T variants in the PMM2 gene for affected individuals and normal siblings is shown in Figure 1B. In-silico prediction of the identified variants in the PMM2 gene is illustrated in Table 1. The c.647A > T (p.N216I) variant was analyzed using multiple in-silico tools, revealing a consensus toward pathogenicity across most algorithms. MetaLR, MetaRNN, and MutPred classified the variant as pathogenic with strong confidence. REVEL and FATHMM provided moderate support for pathogenicity, with scores exceeding commonly accepted pathogenicity thresholds. CADD, with a high score of 29.5, further supports the potential deleterious nature of this variant, as values above 20 are indicative of functional impact. SIFT and LRT classified the variant as deleterious, providing additional supporting evidence. Figure 223 shows Asn 216 within the protein structure. The substitution of asparagine with isoleucine at position 216 likely disrupts crucial hydrogen bonds within the protein, as asparagine possesses an amide group capable of forming hydrogen bonds, while isoleucine is hydrophobic and lacks this ability. The in-silico predictions for the c.640-9T > A variant suggest a high likelihood of pathogenicity. dbscSNV/SpliceAI: Classified as “Deleterious” and “Splice altering Strong,” respectively, indicating a strong potential for this variant to disrupt the splicing process. With a score of 15.77, CADD predicts this variant to be “Possibly Damaging.” While not as strong as the splicing predictions, this score still suggests a significant potential impact on gene function.
Table 1.
In-Silico Prediction of Identified Variants in the PMM2 Gene
|
HGVS c. |
FATHMM |
MetaLR |
SIFT |
REVEL
|
MetaRNN |
MutPred
|
PrimateAI |
LRT |
Mutationtaster
|
dbscSNV/Splice AI
|
CADD
|
| c.640-9T > A |
- |
- |
- |
- |
- |
- |
- |
- |
- |
Deleterious/ splice altering strong |
15.77 |
| c.647A > T |
Pathogenic moderate
(-6.29) |
Pathogenic strong 0.9895 |
Pathogenic supporting
(0) |
Pathogenic moderate 0.927 |
Pathogenic strong
0.9949 |
Pathogenic strong 0.972 |
VUS 0.5389 |
Pathogenic supporting
(0) |
VUS 1 |
- |
29.5 |
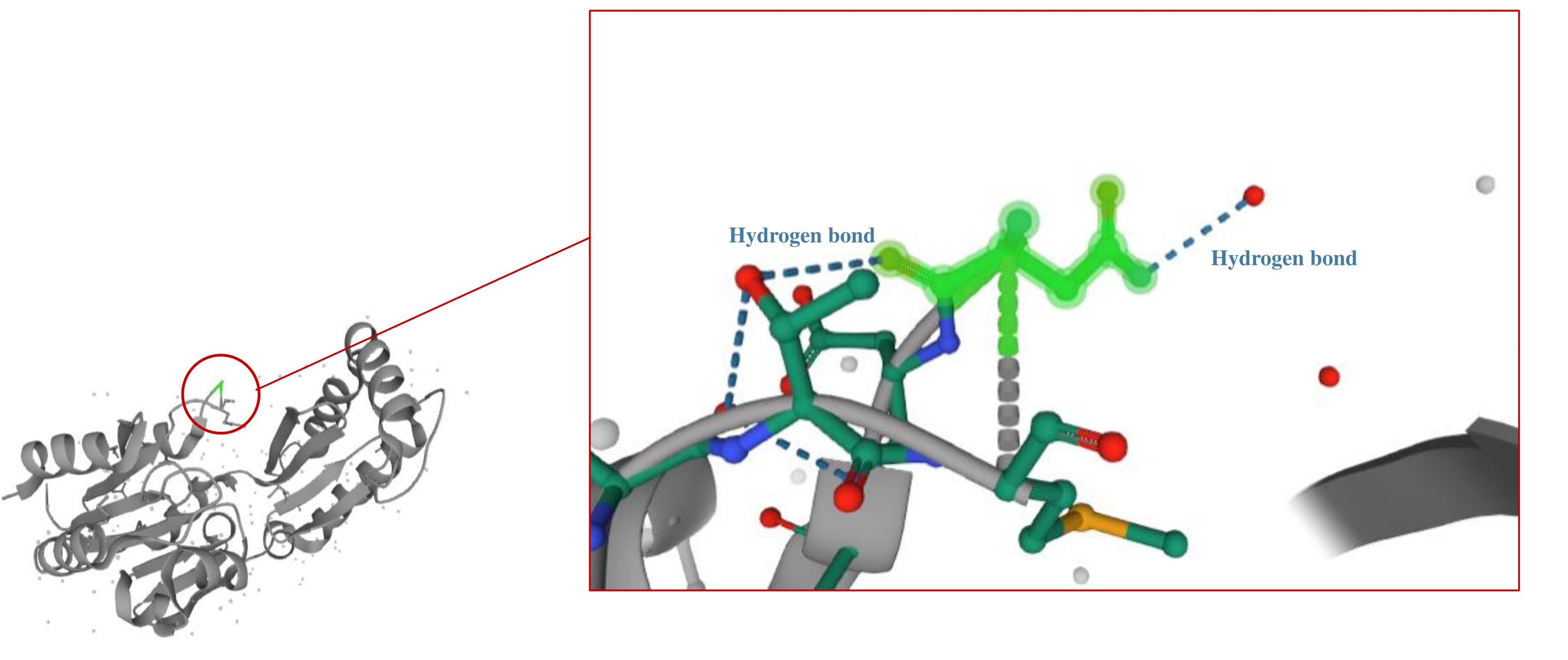
Figure 2.
Asn216 in PMM2 Protein. Substitution of asparagine with isoleucine at position 216 likely disrupts crucial hydrogen bonds within the protein structure, as asparagine can form hydrogen bonds while isoleucine is hydrophobic
.
Asn216 in PMM2 Protein. Substitution of asparagine with isoleucine at position 216 likely disrupts crucial hydrogen bonds within the protein structure, as asparagine can form hydrogen bonds while isoleucine is hydrophobic
Literature Review
Other reports of PMM2-CDG in Iran
According to a study by Piedade et al, no type of CDG is common in multiple countries with high rates of consanguinity, including Iran.24 In Iran, a Middle Eastern country with a parental consanguinity rate of approximately 40%,25 only three families with PMM2-CDG have been reported, including a previously reported family in 201126 by our group, a family reported by Madani et al in 2021 and the present study. In 2011, a missense mutation, p.Y106F, in the PMM2 gene was identified in a consanguineous Iranian family from the Lorestan Province with three affected children presenting with mild intellectual disability, a thin upper lip, a flat nasal bridge, and strabismus. Madani et al identified the p.G117C variant in a patient born to consanguineous parents, who presented with severe hypotonia, motor developmental delay, and elevated urinary 2-ketoglutaric acid levels.27
Mutational Spectrum among PMM2-CDG Patients
According to the Human Gene Mutation Database (HGMD® Professional 2023.4),28 approximately 158 disease-causing PMM2 mutations have been reported so far, with a predominance of synonymous variants. We further reviewed published studies (2017‒2024) reporting PMM2 mutations and associated clinical data. Figures 3A and 3B illustrate these variants at both the protein and genomic levels (illustrated by proteinpaint: https://proteinpaint.stjude.org/).29
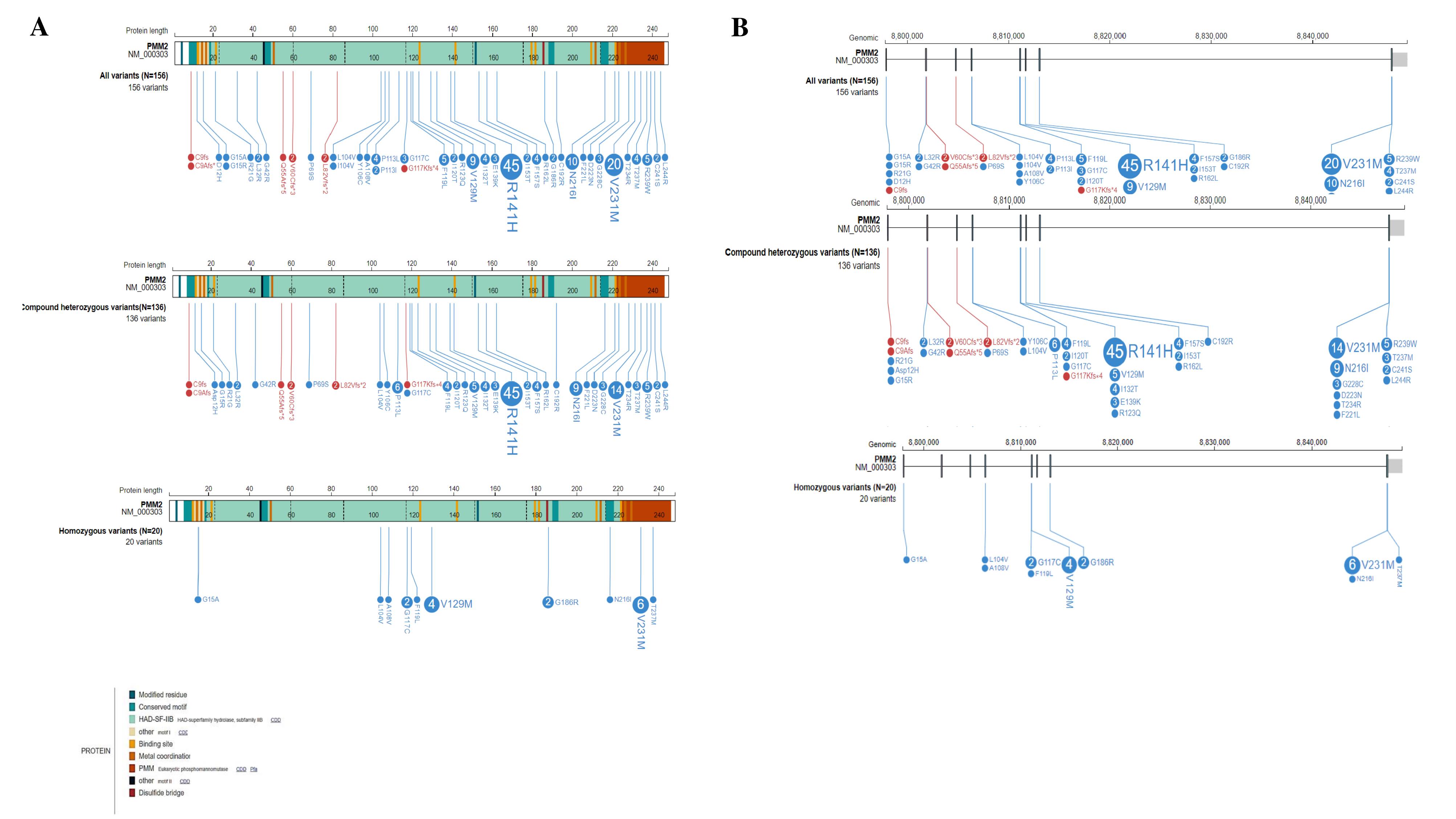
Figure 3.
(A) PMM2 Variants at Protein Level. (B) PMM2 variants at genomic level
.
(A) PMM2 Variants at Protein Level. (B) PMM2 variants at genomic level
Among 91 patients, 76.9% (n = 70) exhibited compound heterozygosity, while 23.1% (n = 21) demonstrated homozygosity. Further calculations were performed on a per-patient basis. Each heterozygous variant was counted once per patient (among 70 patients, 140 alleles were analyzed). For four patients, only one allele with a specified effect on protein sequence was considered. Each homozygous variant was counted once per individual. One patient carrying a homozygous variant, g.18313A > T, was excluded from further analysis due to the complex impact of the variant on the amino acid sequence of the PMM2 protein.30 Altogether, 156 variants were considered, including 20 (12.8%) homozygous and 136 (87.2%) compound heterozygous variants. The majority of variants were missense (94.9%, N = 148), while a smaller proportion were frameshift (5.1%, N = 8). Notably, no frameshift variants were observed among homozygous mutations. The distribution of variants across the exons of the PMM2 gene is shown in Figure 4, revealing that recurrent variants are concentrated in exons 5 and 8, respectively.
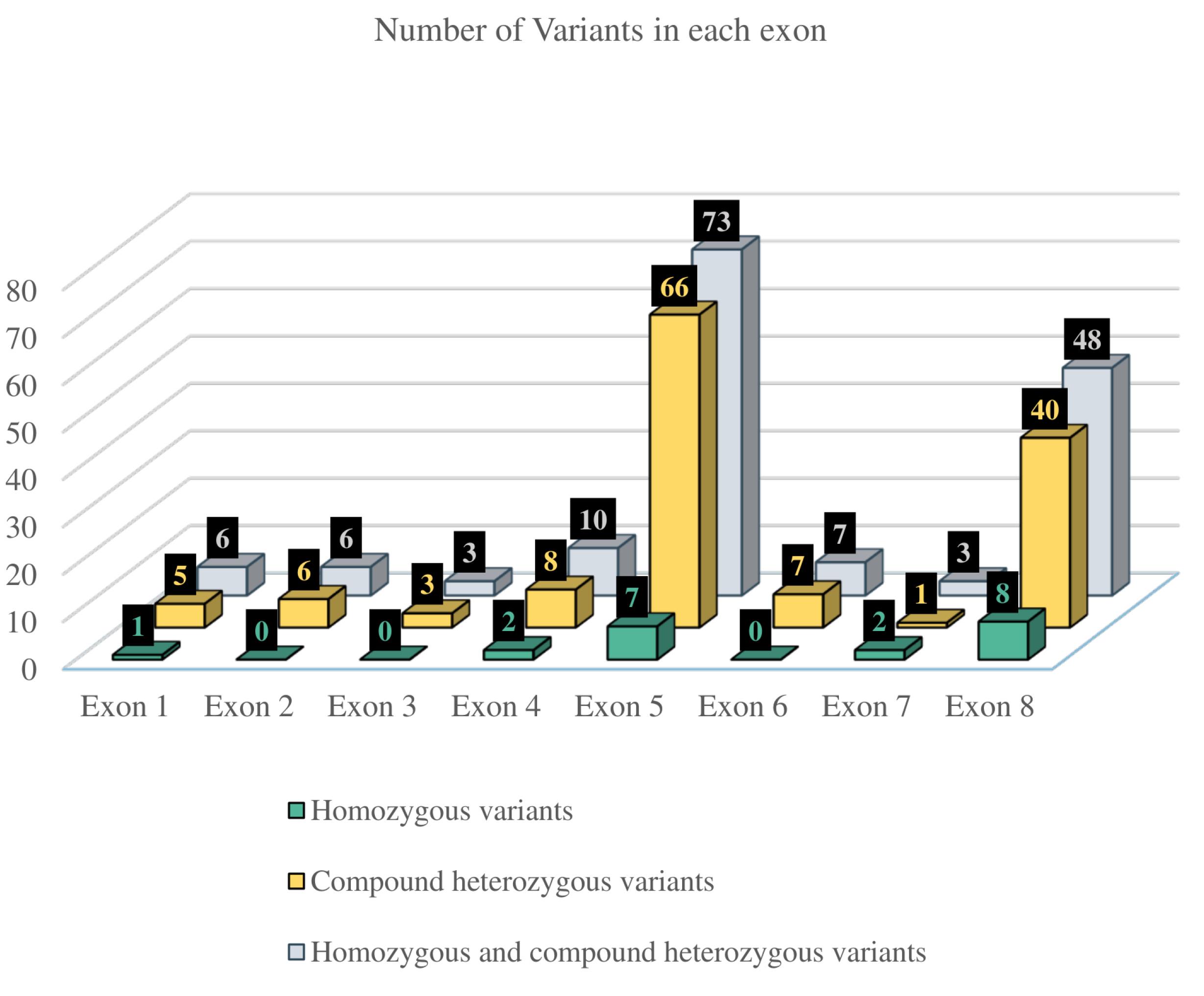
Figure 4.
Distribution of Variants Across the Exons of the PMM2 Gene
.
Distribution of Variants Across the Exons of the PMM2 Gene
Clinical Categorization of 91 Patients
Clinical data were categorized based on the affected body systems, with the results illustrated in Table 2. Developmental delay was one of the most prevalent findings, affecting 87.9% of patients. Ocular abnormalities were common, with hypertelorism (93.3%) and strabismus (70.3%) being the most frequently observed. While a variety of skeletal and skin abnormalities were identified, their prevalence was relatively low. The most common skeletal and skin abnormality was short stature (16.4%), followed by abnormal fat pads (13.2%). Muscular system involvement was primarily characterized by hypotonia or muscle weakness, affecting 70.3% of patients. Neurological manifestations were diverse, with abnormal MRI findings being the most common. Cardiovascular abnormalities were also observed, with pericardial effusion or pericarditis being the most frequent (22%). Hepatic involvement was primarily characterized by elevated transaminases (38.5%). Finally, clotting disorders were common among PMM2-CDG patients, with antithrombin III deficiency being the most prevalent.
Table 2.
Clinical Characterization of Previous Studies Reporting PMM2-CDG Cases With PMM2 Variants
7,27,30-56
|
Organs/clinical signs
|
Clinical Features Reported in 3 or More Cases
|
Clinical Features Reported in<3 Cases (<3.30%)
|
| General |
|
Failure to thrive (27.5%) |
|
| Developmental delay (87.9%) |
| Head & Face |
Head |
Microcephaly (28.6%) |
Macrocephaly |
| Eye |
Strabismus (70.3%) |
Abnormal eyebrows |
| Retinitis pigmentosa (19.8%) |
Abnormal eyelashes |
| Nystagmus (4.4%) |
Retinal dystrophy |
| Hypertelorism (93.3%) |
Eye light sensitivity |
| Abnormal eye movement (3.3%) |
Optic nerve atrophy |
|
|
Macular hypoplasia |
|
|
Decreased visual acuity |
|
|
Wrinkling of the macular retinal surface |
|
|
Cortical visual impairment |
| Ear & Mouth |
Hearing problem (16.5%) |
Postnatal macrosomia |
| Nose |
|
Flat nasal bridge |
| Prominent nares |
| No reaction on nose |
| Wide nasal bridge |
| Skeletal system |
Osteoporosis/ Osteopenia (4.4%) |
Kyphoscoliosis |
| Scoliosis (7/9%) |
Clubfoot |
| Joint laxity (4.4%) |
Bilateral radial aplasia |
| Short stature (16.5%) |
Pectus Carinatum |
|
|
Pectus excavatum |
| Scapular dyskinesis (Mild winging of the scapulae) |
| Talipes equinus |
| Hammer toe |
| Elongated slender fingers |
| Spinal cord disorder |
| Skin |
Abnormal fat distribution (13.2%) |
Purpura |
| Orange peel' skin (7.7%) |
Petechiae |
|
|
Pressure ulcers |
| pilonidal sinus, |
| Skin elasticity changes |
| Easy bruising |
| Eczema |
| Dry skin parts |
| Muscular system |
Muscle weakness or hypotonia (70.3%) |
Spasticity |
| Myopathy (15.4%) |
Torticollis |
|
|
Tendon Reflexes and Plantar Responses |
| Spastic paraplegia |
| Nervous system |
Ataxia (28.6%) |
Non-cerebral haemorrhage |
| Hyporeflexia (14.3%) |
Intentional tremor |
| Stroke-like episodes (14.3%) |
|
| Seizure or epilepsy (27.5%) |
|
| Stroke mimic (3.3%) |
|
| Cerebral thrombosis (5.5%) |
|
| Non-cerebral thrombosis (3.3%) |
|
| Cerebral haemorrhage (4.4%) |
|
| Abnormal MRI results (68.1%) |
|
| Peripheral neuropathy (16.5%) |
|
| Urogenital system |
Genital |
POF or risk of POF (5.5%) |
Defect in secondary sexual development |
| Urinary system |
Proteinuria (16.5%) |
Nephrocalcinosis |
| Increased renal echogenicity (15.4%) |
Renal cyst/ cystic renal disorder |
| Tubulopathy (5.5%) |
Oliguria |
| Cryptorchidism (3.3%) |
Hypertension due to nephrotic syndrome |
| Inguinal hernia (3.3%) |
Enlarged kidney and decreased corticomedullary diameter |
| Cardiovascular system |
Pericardial effusion/pericarditis (22%) |
Conotruncal cardiac malformations |
| Liver problems |
|
Hepatomegaly (18.7%) |
Liver fibrosis |
|
|
|
Increased liver echogenicity (12.1%) |
Steatosis |
|
|
|
Elevated transaminases (38.5%) |
Liver failure |
|
|
|
|
Low haptoglobin level |
| Other signs |
Nipple anomalies (38.5%) |
Headache |
| Ascites (11%) |
Steatorrhea |
| Edema (7.7%) |
|
| Behaviour changes (3.3%) |
|
| Gastrointstinal problems |
Feeding problems (22%) |
Gastroesophageal reflux diseas |
|
|
Vomiting (10%) |
|
|
|
Diarrhea (10%) |
|
| Respiratory system |
Pneumonia (5.5%) |
Sleep apnea |
| Pleural effusion (6.6%) |
Episodes of cyanosis |
|
|
Sinusitis |
| Pulmonary nodular amyloidosis |
| Tachypnea |
| Dyspnea |
| Recurrent airway infections |
| Hypoxemia |
| Bronchopneumonia |
| Respiratory distress |
| Meconium aspiration syndrome |
| Endocrine system |
Hypothyroidism (16.5%) |
Thyroid binding globulin deficiency |
| Hypergonadotropic hypogonadism (4.4%) |
Hyperprolactinemia |
| Hyperinsulinaemic hypoglycaemia (12.1%) |
Panhypopituitarism/hypoplastic infundibulum |
| Adrenal insufficiency (3.3%) |
Hypomagnesemia |
| GH deficiency |
| Prenatal Manifestations |
Non-immune hydrops fetalis (3.3%) |
Low birth weight |
|
|
Oligohydramnios |
| Intrauterine growth retardation |
| Biochemistry |
Hypocholesterolemia (4.4%) |
Hypolipidaemia |
| Triglyceridemia (9. 9%) |
Hyperammonemia |
| Hypoalbuminemia (12.1%) |
Lactic acidosis |
| Low serum HDL (4.4%) |
Iron deficiency |
|
|
Abnormal ferritin levels |
|
|
High 2-ketoglutaric acid in urine sample |
|
|
Low circulating PCSK9 levels |
|
|
Hypoproteinemia |
|
|
Elevation of dehydrogenase |
|
|
Low microalbumin |
|
|
Low serum creatinin |
|
|
High serum creatinin |
|
|
Elevation of creatine kinase |
|
|
Low urine beta-2 microglobulin |
| Immunology |
Hypogammaglobulinemia (3.3%) |
Leucocytosis (high wbc) |
| Hematology |
Anemia (5.5%) |
Low INR |
| Thrombocytopenia (5.5%) |
Pancytopenia |
| Thrombocytosis (3.3%) |
Low factor X |
| High INR (5.5%) |
High factor VIII |
| Prolonged PT (9.9%) |
|
| Factor XI deficiency (14.3%) |
|
| Antithrombin III deficiency (34.1%) |
|
| Low protein C (9.9%) |
|
| Low protein S (7.7%) |
|
| Low factor XI (9.9%) |
|
| Low factor IX (4.4%) |
|
Discussion
Our family harbors a previously reported missense mutation (p.N216I) and a novel intronic splice site variant (c.640-9T > A). Since 1997, multiple studies have reported the p.N216I allele in a compound heterozygous state with the p.R141H allele in Italian patients.57,58 The only reported homozygous genotype for this variant was observed in an unusual case of PMM2-CDG, presenting with postnatal macrosomia, distinctive bushy eyebrows with an abnormally shaped right eyebrow, and an absence of inverted nipples and fat pads. Notably, motor nerve conduction velocity in the tibialis posterior nerve of the lower limbs was normal, in contrast to other PMM2-CDG patients.59 As depicted in Figure 2,23 asparagine’s side chain contains an amide group (-CONH2), capable of forming hydrogen bonds as both a donor and acceptor. In contrast, isoleucine is a hydrophobic amino acid with a hydrocarbon side chain, unable to participate in hydrogen bonding. Thus, the substitution of asparagine with isoleucine at position 216 likely disrupts crucial protein interactions and hydrogen bonding, contributing to the observed phenotypic features in PMM2-CDG patients carrying this mutation. The other identified variant, which is a novel variant, c.640-9T > A, is located within the polypyrimidine tract of the last intron of the PMM2 gene. To the authors’ knowledge, this variant has not been reported before, but multiple reports of intronic variant NM_000303.3:c.640-9T > G exist. A previous study identified this variant as important for the activation of a cryptic intronic splice site in fibroblast cell lines.60
The PMM2-CDG is a rare disorder with only three reported families in Iran, of which two were identified by our group. The identification of recurrent variants, particularly p.R141H, p.V231M, p.N216I, and p.V129M, highlights the importance of these specific alterations in disease pathogenesis. The clustering of variants in exons 5 and 8 suggests potential mutational hotspots that may be targeted for efficient genetic testing. The absence of synonymous variants within the two conserved domains (amino acids 46‒48 and 188‒190) of the PMM2 gene (Figure 3) may suggest that these regions are highly conserved and critical for the protein’s function. However, further studies are still needed to investigate this issue.
As this is a descriptive study, we conducted a comprehensive review of published reports on PMM2 variants and their associated clinical features. Common clinical findings included developmental delay, ocular problems (hypertelorism and strabismus), muscular system abnormalities (hypotonia or muscle weakness), neurological signs (abnormal MRI findings), cardiovascular system involvement (pericarditis or pericardial effusion), hepatic problems (elevated transaminases) and clotting disorders (antithrombin III deficiency). Less common findings were skeletal and skin abnormalities, and behavioral problems. This study showed that prenatal manifestations are rare among PMM2-CDG patients but they include non-immune hydrops fetalis, low birth weight, oligohydramnios and intrauterine growth retardation.
Conclusion
In conclusion, our study reports a novel splice variant with a nucleotide substitution in a family with PMM2-CDG and expands the knowledge on PMM2-CDG by reviewing 91 previously reported cases. The most prevalent variants and recurrent mutations occurred in exons 5 and 8 of the PMM2 gene. A limitation of this study is that the categorization of papers was based solely on clinical signs explicitly stated by authors. This approach may have inadvertently excluded some clinical signs that, while present, were not explicitly mentioned or investigated. Furthermore, the inclusion criteria of studies reporting both genotype and phenotype data could introduce bias, as not all relevant studies may meet this specific requirement. Since the incidence of PMM2-CDG is approximately 1 in 20 000,61 our analysis of 91 well-documented cases—each reporting both clinical and genetic data—represents a substantial portion of the fully reported cases currently available in the literature. This allowed us to explore clinical sign classifications and identify potential genetic hotspots. However, it should be noted that not all reported cases could be included, particularly those lacking comprehensive individual-level clinical or genetic information. To mitigate these limitations and draw more robust conclusions, a comprehensive analysis of phenotypic and clinical data from a broader range of studies is necessary. For this reason, authors advocate for the creation of a comprehensive database containing both clinical and genotype data of PMM2-CDG patients.
Acknowledgements
We are grateful to the family for their participation in this study. We also extend our sincere thanks to everyone who contributed to this project. This research would not have been possible without their generous support and assistance.
Competing Interests
The authors declare no conflict of interest.
Declaration of Generative AI and AI-assisted Technologies in the Writing Process
During the preparation of this work, the authors used ChatGPT and Gemini for language refinement and grammatical corrections. After using these tools, the authors reviewed and edited the content as needed and take full responsibility for the content of the published article.
Ethical Approval
The study was approved by the Ethics Committee of the University of Social Welfare and Rehabilitation Sciences, Tehran, Iran (Ethics approval No.:IR.USWR.REC.1403.044). Written informed consent was obtained from the parents of the patients and normal siblings.
Funding
This project was supported by the Iran National Science Foundation (INSF) under Grant No. 96011200 to Kimia Kahrizi (KK) and grant numbers of 1402/801/A/6/28595 and 03/T/308 to Hossein Najmabadi.2
References
- Schiff M, Roda C, Monin ML, Arion A, Barth M, Bednarek N. Clinical, laboratory and molecular findings and long-term follow-up data in 96 French patients with PMM2-CDG (phosphomannomutase 2-congenital disorder of glycosylation) and review of the literature. J Med Genet 2017; 54(12):843-51. doi: 10.1136/jmedgenet-2017-104903 [Crossref] [ Google Scholar]
- Muthusamy K, Perez-Ortiz JM, Ligezka AN, Altassan R, Johnsen C, Schultz MJ. Neurological manifestations in PMM2-congenital disorders of glycosylation (PMM2-CDG): insights into clinico-radiological characteristics, recommendations for follow-up, and future directions. Genet Med 2024; 26(2):101027. doi: 10.1016/j.gim.2023.101027 [Crossref] [ Google Scholar]
- Chang IJ, He M, Lam CT. Congenital disorders of glycosylation. Ann Transl Med 2018; 6(24):477. doi: 10.21037/atm.2018.10.45 [Crossref] [ Google Scholar]
- Pettinato F, Mostile G, Battini R, Martinelli D, Madeo A, Biamino E. Clinical and radiological correlates of activities of daily living in cerebellar atrophy caused by PMM2 mutations (PMM2-CDG). Cerebellum 2021; 20(4):596-605. doi: 10.1007/s12311-021-01242-x [Crossref] [ Google Scholar]
- Altassan R, Péanne R, Jaeken J, Barone R, Bidet M, Borgel D. International clinical guidelines for the management of phosphomannomutase 2-congenital disorders of glycosylation: diagnosis, treatment and follow up. J Inherit Metab Dis 2019; 42(1):5-28. doi: 10.1002/jimd.12024 [Crossref] [ Google Scholar]
- Vilas A, Yuste-Checa P, Gallego D, Desviat LR, Ugarte M, Pérez-Cerda C. Proteostasis regulators as potential rescuers of PMM2 activity. Biochim Biophys Acta Mol Basis Dis 2020; 1866(7):165777. doi: 10.1016/j.bbadis.2020.165777 [Crossref] [ Google Scholar]
- Vals MA, Morava E, Teeäär K, Zordania R, Pajusalu S, Lefeber DJ. Three families with mild PMM2-CDG and normal cognitive development. Am J Med Genet A 2017; 173(6):1620-4. doi: 10.1002/ajmg.a.38235 [Crossref] [ Google Scholar]
- Epifani F, Pujol Serra SM, Llorens M, Balcells S, Nolasco G, Bolasell M. Untangling adaptive functioning of PMM2-CDG across age and its impact on parental stress: a cross-sectional study. Sci Rep 2023; 13(1):22783. doi: 10.1038/s41598-023-49518-y [Crossref] [ Google Scholar]
- Lam C, Krasnewich DM. PMM2-CDG. In: GeneReviews®. Seattle, WA: University of Washington, Seattle; 1993.
- Quental R, Moleirinho A, Azevedo L, Amorim A. Evolutionary history and functional diversification of phosphomannomutase genes. J Mol Evol 2010; 71(2):119-27. doi: 10.1007/s00239-010-9368-5 [Crossref] [ Google Scholar]
- Wang J, Chitsaz F, Derbyshire MK, Gonzales NR, Gwadz M, Lu S. The conserved domain database in 2023. Nucleic Acids Res 2023; 51(D1):D384-8. doi: 10.1093/nar/gkac1096 [Crossref] [ Google Scholar]
- Silvaggi NR, Zhang C, Lu Z, Dai J, Dunaway-Mariano D, Allen KN. The X-ray crystal structures of human alpha-phosphomannomutase 1 reveal the structural basis of congenital disorder of glycosylation type 1a. J Biol Chem 2006; 281(21):14918-26. doi: 10.1074/jbc.M601505200 [Crossref] [ Google Scholar]
- Zhong D, Huang X, Feng T, Zeng J, Gu S, Ning F. Congenital disorder of glycosylation type Ia in a Chinese family: function analysis of a novel PMM2 complex heterozygosis mutation. Mol Genet Metab Rep 2024; 39:101067. doi: 10.1016/j.ymgmr.2024.101067 [Crossref] [ Google Scholar]
- Gámez A, Serrano M, Gallego D, Vilas A, Pérez B. New and potential strategies for the treatment of PMM2-CDG. Biochim Biophys Acta Gen Subj 2020; 1864(11):129686. doi: 10.1016/j.bbagen.2020.129686 [Crossref] [ Google Scholar]
- Himmelreich N, Kikul F, Zdrazilova L, Honzik T, Hecker A, Poschet G. Complex metabolic disharmony in PMM2-CDG paves the way to new therapeutic approaches. Mol Genet Metab 2023; 139(3):107610. doi: 10.1016/j.ymgme.2023.107610 [Crossref] [ Google Scholar]
- Taday R, Grüneberg M, DuChesne I, Reunert J, Marquardt T. Dietary mannose supplementation in phosphomannomutase 2 deficiency (PMM2-CDG). Orphanet J Rare Dis 2020; 15(1):258. doi: 10.1186/s13023-020-01528-z [Crossref] [ Google Scholar]
- Medina-Cano D, Ucuncu E, Nguyen LS, Nicouleau M, Lipecka J, Bizot JC. High N-glycan multiplicity is critical for neuronal adhesion and sensitizes the developing cerebellum to N-glycosylation defect. Elife 2018; 7:e38309. doi: 10.7554/eLife.38309 [Crossref] [ Google Scholar]
- Mutalik SP, Gupton SL. Glycosylation in axonal guidance. Int J Mol Sci 2021; 22(10):5143. doi: 10.3390/ijms22105143 [Crossref] [ Google Scholar]
- Thiel C, Lübke T, Matthijs G, von Figura K, Körner C. Targeted disruption of the mouse phosphomannomutase 2 gene causes early embryonic lethality. Mol Cell Biol 2006; 26(15):5615-20. doi: 10.1128/mcb.02391-05 [Crossref] [ Google Scholar]
- Klarić TS, Lauc G. The dynamic brain N-glycome. Glycoconj J 2022; 39(3):443-71. doi: 10.1007/s10719-022-10055-x [Crossref] [ Google Scholar]
- Scheper AF, Schofield J, Bohara R, Ritter T, Pandit A. Understanding glycosylation: regulation through the metabolic flux of precursor pathways. Biotechnol Adv 2023; 67:108184. doi: 10.1016/j.biotechadv.2023.108184 [Crossref] [ Google Scholar]
- Tosh N, Quadrelli S, Galloway G, Mountford C. Two new fucose-α (1-2)-glycans assigned in the healthy human brain taking the number to seven. Sci Rep 2019; 9(1):18806. doi: 10.1038/s41598-019-54933-1 [Crossref] [ Google Scholar]
- UniProt Consortium. UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res 2023; 51(D1):D523-31. doi: 10.1093/nar/gkac1052 [Crossref] [ Google Scholar]
- Piedade A, Francisco R, Jaeken J, Sarkhail P, Brasil S, Ferreira CR. Epidemiology of congenital disorders of glycosylation (CDG)—overview and perspectives. J Rare Dis (Berlin) 2022; 1(1):3. doi: 10.1007/s44162-022-00003-6 [Crossref] [ Google Scholar]
- Zare Ashrafi F, Akhtarkhavari T, Fattahi Z, Asadnezhad M, Beheshtian M, Arzhangi S. Emerging epidemiological data on rare intellectual disability syndromes from analyzing the data of a large Iranian cohort. Arch Iran Med 2023; 26(4):186-97. doi: 10.34172/aim.2023.29 [Crossref] [ Google Scholar]
- Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W. Deep sequencing reveals 50 novel genes for recessive cognitive disorders. Nature 2011; 478(7367):57-63. doi: 10.1038/nature10423 [Crossref] [ Google Scholar]
- Madani S, Sayarifard F, Tajdini P, Mohsenipour R, Khoram Khorshid HR, Rezaei N. Novel treatment for congenital disorder of glycosylation in a patient with novel homozygote mutation of PMM2: a case report and review literature. Endocr Metab Immune Disord Drug Targets 2021; 21(12):2296-9. doi: 10.2174/1871530321666210415105917 [Crossref] [ Google Scholar]
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet 2020; 139(10):1197-207. doi: 10.1007/s00439-020-02199-3 [Crossref] [ Google Scholar]
- Zhou X, Edmonson MN, Wilkinson MR, Patel A, Wu G, Liu Y. Exploring genomic alteration in pediatric cancer using ProteinPaint. Nat Genet 2016; 48(1):4-6. doi: 10.1038/ng.3466 [Crossref] [ Google Scholar]
- Görlacher M, Panagiotou E, Himmelreich N, Hüllen A, Beedgen L, Dimitrov B. Fatal outcome after heart surgery in PMM2-CDG due to a rare homozygous gene variant with double effects. Mol Genet Metab Rep 2020; 25:100673. doi: 10.1016/j.ymgmr.2020.100673 [Crossref] [ Google Scholar]
- Wicker C, Roux CJ, Goujon L, de Feraudy Y, Hully M, Brassier A. Association between acute complications in PMM2-CDG patients and haemostasis anomalies: data from a multicentric study and suggestions for acute management. Mol Genet Metab 2023; 140(3):107674. doi: 10.1016/j.ymgme.2023.107674 [Crossref] [ Google Scholar]
- Raynor A, Bruneel A, Vermeersch P, Cholet S, Friedrich S, Eckenweiler M. “Hide and seek”: misleading transferrin variants in PMM2-CDG complicate diagnostics. Proteomics Clin Appl 2024; 18(2):e2300040. doi: 10.1002/prca.202300040 [Crossref] [ Google Scholar]
- Hong X, Edmondson AC, Strong A, Pomerantz D, Michl E, Berry G. Combined PMM2-CDG and hereditary fructose intolerance in a patient with mild clinical presentation. Mol Genet Metab 2023; 140(3):107682. doi: 10.1016/j.ymgme.2023.107682 [Crossref] [ Google Scholar]
- Cirnigliaro L, Pettinato F, Valle MS, Casabona A, Fiumara A, Vecchio M. Instrumented assessment of gait disturbance in PMM2-CDG adults: a feasibility analysis. Orphanet J Rare Dis 2024; 19(1):39. doi: 10.1186/s13023-024-03027-x [Crossref] [ Google Scholar]
- Asteggiano CG, Papazoglu M, Bistué Millón MB, Peralta MF, Azar NB, Spécola NS. Ten years of screening for congenital disorders of glycosylation in Argentina: case studies and pitfalls. Pediatr Res 2018; 84(6):837-41. doi: 10.1038/s41390-018-0206-6 [Crossref] [ Google Scholar]
- Farmania R, Jain P, Sharma S, Aneja S. Unusual presentation of PMM2-congenital disorder of glycosylation with isolated strokelike episodes in a young girl. J Child Neurol 2019; 34(7):410-4. doi: 10.1177/0883073819833543 [Crossref] [ Google Scholar]
- Peng T, Lv C, Tan H, Huang J, He H, Wang Y. Novel PMM2 missense mutation in a Chinese family with non-syndromic premature ovarian insufficiency. J Assist Reprod Genet 2020; 37(2):443-50. doi: 10.1007/s10815-019-01675-8 [Crossref] [ Google Scholar]
- Qian Z, Van den Eynde J, Heymans S, Mertens L, Morava E. Vascular ring anomaly in a patient with phosphomannomutase 2 deficiency: a case report and review of the literature. JIMD Rep 2020; 56(1):27-33. doi: 10.1002/jmd2.12160 [Crossref] [ Google Scholar]
- Grünert SC, Marquardt T, Lausch E, Fuchs H, Thiel C, Sutter M. Unsuccessful intravenous D-mannose treatment in PMM2-CDG. Orphanet J Rare Dis 2019; 14(1):231. doi: 10.1186/s13023-019-1213-3 [Crossref] [ Google Scholar]
- Kasapkara Ç S, Barış Z, Kılıç M, Yüksel D, Keldermans L, Matthijs G. PMM2-CDG and sensorineural hearing loss. J Inherit Metab Dis 2017; 40(5):629-30. doi: 10.1007/s10545-017-0073-z [Crossref] [ Google Scholar]
- Chong M, Yoon G, Susan-Resiga D, Chamberland A, Cheillan D, Paré G. Hypolipidaemia among patients with PMM2-CDG is associated with low circulating PCSK9 levels: a case report followed by observational and experimental studies. J Med Genet 2020; 57(1):11-7. doi: 10.1136/jmedgenet-2019-106102 [Crossref] [ Google Scholar]
- Lefrère B, Stepanian A, Charles P, Foulon-Pinto G, Béranger N, Alhenc-Gelas M. Multifactorial hypercoagulable state associated with a thrombotic phenotype in phosphomannomutase-2 congenital disorder of glycosylation (PMM2-CDG): case report and brief review of the literature. Thromb Res 2019; 178:75-8. doi: 10.1016/j.thromres.2019.04.010 [Crossref] [ Google Scholar]
- Van Hees I, Jaeken J, Meersseman W, Casteels I. Ophthalmological and electrophysiological findings in monozygotic twin sisters with phosphomannomutase 2 deficiency (PMM2-CDG) over a period of 37 years. GMS Ophthalmol Cases 2019; 9:Doc37. doi: 10.3205/oc000126 [Crossref] [ Google Scholar]
- Zhang Z, Huang TL, Ma J, He WJ, Gu H. Clinical and whole-exome sequencing findings in two siblings from Hani ethnic minority with congenital glycosylation disorders. BMC Med Genet 2019; 20(1):181. doi: 10.1186/s12881-019-0902-z [Crossref] [ Google Scholar]
- Vaes L, Tiller GE, Pérez B, Boyer SW, Berry SA, Sarafoglou K. PMM2-CDG caused by uniparental disomy: case report and literature review. JIMD Rep 2020; 54(1):16-21. doi: 10.1002/jmd2.12122 [Crossref] [ Google Scholar]
- Masunaga Y, Mochizuki M, Kadoya M, Wada Y, Okamoto N, Fukami M. Primary ovarian insufficiency in a female with phosphomannomutase-2 gene (PMM2) mutations for congenital disorder of glycosylation. Endocr J 2021; 68(5):605-11. doi: 10.1507/endocrj.EJ20-0706 [Crossref] [ Google Scholar]
- Lipiński P, Cielecka-Kuszyk J, Czarnowska E, Bogdańska A, Socha P, Tylki-Szymańska A. Congenital disorders of glycosylation in children - histopathological and ultrastructural changes in the liver. Pediatr Neonatol 2021; 62(3):278-83. doi: 10.1016/j.pedneo.2021.01.017 [Crossref] [ Google Scholar]
- Yıldız Y, Arslan M, Çelik G, Kasapkara ÇS, Ceylaner S, Dursun A. Genotypes and estimated prevalence of phosphomannomutase 2 deficiency in Turkey differ significantly from those in Europe. Am J Med Genet A 2020; 182(4):705-12. doi: 10.1002/ajmg.a.61488 [Crossref] [ Google Scholar]
- Tiwary H, Hecht LE, Brucker WJ, Berry GT, Rodig NM. The development of end stage renal disease in two patients with PMM2-CDG. JIMD Rep 2022; 63(2):131-6. doi: 10.1002/jmd2.12269 [Crossref] [ Google Scholar]
- González-Domínguez CA, Villarroel CE, Rodríguez-Morales M, Manrique-Hernández S, González-Jaimes A, Olvera-Rodriguez F. Non-functional alternative splicing caused by a Latino pathogenic variant in a case of PMM2-CDG. Mol Genet Metab Rep 2021; 28:100781. doi: 10.1016/j.ymgmr.2021.100781 [Crossref] [ Google Scholar]
- González-Domínguez CA, Raya-Trigueros A, Manrique-Hernández S, González Jaimes A, Salinas-Marín R, Molina-Garay C. Identification through exome sequencing of the first PMM2-CDG individual of Mexican mestizo origin. Mol Genet Metab Rep 2020; 25:100637. doi: 10.1016/j.ymgmr.2020.100637 [Crossref] [ Google Scholar]
- Sreedevi N, Swapna N, Maruthy S, Meghavathi HS, Sylvester C. PMM2 -CDG T237M mutation in a patient with cerebral palsy-like phenotypes reported from South India. Glob Med Genet 2023; 10(2):105-8. doi: 10.1055/s-0043-1769494 [Crossref] [ Google Scholar]
- Tahata S, Weckwerth J, Ligezka A, He M, Lee HE, Heimbach J. Liver transplantation recovers hepatic N-glycosylation with persistent IgG glycosylation abnormalities: three-year follow-up in a patient with phosphomannomutase-2-congenital disorder of glycosylation. Mol Genet Metab 2023; 138(4):107559. doi: 10.1016/j.ymgme.2023.107559 [Crossref] [ Google Scholar]
- Yoldas Celik M, Yazici H, Erdem F, Yuksel Yanbolu A, Aykut A, Durmaz A. Unique clinical presentations and follow-up outcomes from experience with congenital disorders of glycosylation: PMM2-PGM1-DPAGT1-MPI-POMT2-B3GALNT2-DPM1-SRD5A3-CDG. J Pediatr Endocrinol Metab 2023; 36(6):530-8. doi: 10.1515/jpem-2022-0641 [Crossref] [ Google Scholar]
- Slaba K, Noskova H, Vesela P, Tuckova J, Jicinska H, Honzik T. Novel Splicing Variant in the PMM2 Gene in a Patient With PMM2-CDG Syndrome Presenting With Pericardial Effusion: A Case Report. Front Genet 2020; 11:561054. doi: 10.3389/fgene.2020.561054 [Crossref] [ Google Scholar]
- Zhong M, Balakrishnan B, Guo AJ, Lai K. AAV9-based PMM2 gene replacement augments PMM2 expression and improves glycosylation in primary fibroblasts of patients with phosphomannomutase 2 deficiency (PMM2-CDG). Mol Genet Metab Rep 2024; 38:101035. doi: 10.1016/j.ymgmr.2023.101035 [Crossref] [ Google Scholar]
- Matthijs G, Schollen E, Pardon E, Veiga-Da-Cunha M, Jaeken J, Cassiman JJ. Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome). Nat Genet 1997; 16(1):88-92. doi: 10.1038/ng0597-88 [Crossref] [ Google Scholar]
- Barone R, Carrozzi M, Parini R, Battini R, Martinelli D, Elia M. A nationwide survey of PMM2-CDG in Italy: high frequency of a mild neurological variant associated with the L32R mutation. J Neurol 2015; 262(1):154-64. doi: 10.1007/s00415-014-7549-7 [Crossref] [ Google Scholar]
- Neumann LM, von Moers A, Kunze J, Blankenstein O, Marquardt T. Congenital disorder of glycosylation type 1a in a macrosomic 16-month-old boy with an atypical phenotype and homozygosity of the N216I mutation. Eur J Pediatr 2003; 162(10):710-3. doi: 10.1007/s00431-003-1278-8 [Crossref] [ Google Scholar]
- Vega AI, Pérez-Cerdá C, Desviat LR, Matthijs G, Ugarte M, Pérez B. Functional analysis of three splicing mutations identified in the PMM2 gene: toward a new therapy for congenital disorder of glycosylation type Ia. Hum Mutat 2009; 30(5):795-803. doi: 10.1002/humu.20960 [Crossref] [ Google Scholar]
- Pajusalu S, Vals MA, Mihkla L, Šamarina U, Kahre T, Õunap K. The estimated prevalence of N-linked congenital disorders of glycosylation across various populations based on allele frequencies in general population databases. Front Genet 2021; 12:719437. doi: 10.3389/fgene.2021.719437 [Crossref] [ Google Scholar]